≥99%HPLC Purity
COAIncluded
USAShipped
Ipamorelin Peptide
Selective growth hormone secretagogue pentapeptide. Studied for its GHS-R1a receptor selectivity in published research.
Coming Soon
This compound is currently in our development pipeline. Enter your email to be notified when it becomes available for research purchase.
Best price — free shipping & bac water included
| Simple Peptide | AminoCore ✓ all‑in | |
|---|---|---|
| 10mg price | $65.00 | $55.00 |
| Shipping | +$13.00 | FREE |
| Bac Water | +$30.00 | FREE |
| Total paid | $108.00 | $55.00 save up to $53.00 |
Quick Facts
| SKU | ACR-IPAM |
|---|---|
| CAS Number | 170851-70-4 |
| Molecular Formula | C38H49N9O5 |
| Molecular Weight | 711.85 g/mol |
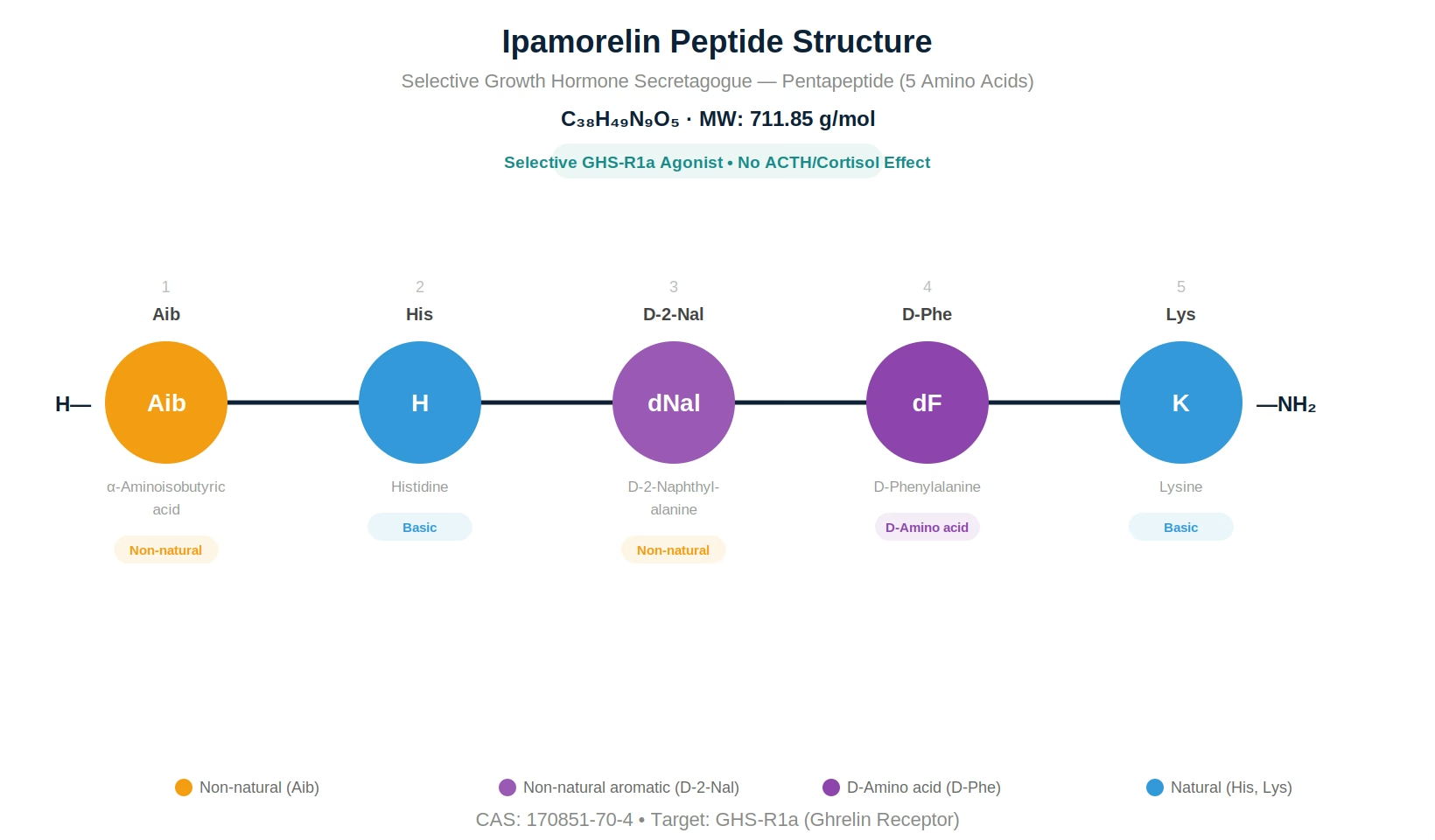
| Sequence | Aib-His-D-2-Nal-D-Phe-Lys-NH2 |
| Purity | ≥98% |
| Physical Form | Lyophilized Powder |
| Storage | Store at -20°C |
What is Ipamorelin?
Ipamorelin is a synthetic pentapeptide growth hormone secretagogue (GHS) with the sequence Aib-His-D-2-Nal-D-Phe-Lys-NH2, where Aib is alpha-aminoisobutyric acid and D-2-Nal is D-2-naphthylalanine. It has a molecular weight of 711.85 g/mol and the CAS registry number 170851-70-4. Ipamorelin was developed by Novo Nordisk in the late 1990s as part of a systematic effort to create growth hormone secretagogues with improved selectivity profiles.
Ipamorelin selectively activates the growth hormone secretagogue receptor type 1a (GHS-R1a), the same receptor targeted by the endogenous hormone ghrelin. However, unlike ghrelin and earlier synthetic GHS compounds such as GHRP-6 and GHRP-2, Ipamorelin demonstrates remarkable selectivity for growth hormone (GH) release without significant stimulation of adrenocorticotropic hormone (ACTH), cortisol, or prolactin — even at doses far exceeding those required for maximal GH secretion.
This selectivity profile was first characterized by Raun et al. (1998) in a landmark study published in the European Journal of Endocrinology, which demonstrated that Ipamorelin produced dose-dependent GH release comparable to GHRP-6 in potency but without the accompanying rises in cortisol and prolactin that characterized all previously known GHS compounds. This finding established Ipamorelin as the first truly selective GH secretagogue.
The pentapeptide incorporates two non-natural amino acids (Aib and D-2-Nal) and one D-configured residue (D-Phe), providing substantial resistance to enzymatic degradation while maintaining optimal GHS-R1a receptor binding conformation. The C-terminal amidation further enhances metabolic stability and receptor affinity.
Ipamorelin is classified as a research peptide and is available exclusively for laboratory and scientific investigation. All findings described herein are derived from published research and do not constitute medical claims.
Mechanism of Action
Published research has characterized Ipamorelin as a selective GHS-R1a agonist with a well-defined signaling cascade that converges on anterior pituitary somatotroph cells.
GHS-R1a Receptor Activation
Ipamorelin binds to the growth hormone secretagogue receptor type 1a (GHS-R1a), a 366-amino acid G-protein coupled receptor predominantly expressed on somatotroph cells of the anterior pituitary gland and in the arcuate nucleus of the hypothalamus. GHS-R1a is a Gq/11-coupled receptor — upon Ipamorelin binding, it activates phospholipase C-beta (PLCβ), generating inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 triggers calcium release from the endoplasmic reticulum, while DAG activates protein kinase C (PKC). The resulting intracellular calcium surge is the proximate trigger for GH vesicle exocytosis from somatotroph cells.
Selectivity for GH Release
The defining pharmacological feature of Ipamorelin is its selectivity for GH release over other pituitary hormones. Raun et al. (1998) demonstrated that at doses producing maximal GH secretion, Ipamorelin did not significantly increase plasma ACTH, cortisol, or prolactin levels. This stands in stark contrast to GHRP-6, which at equipotent GH-releasing doses produces significant rises in all three hormones. The molecular basis for this selectivity is not fully understood but is believed to involve subtle differences in how Ipamorelin engages the GHS-R1a binding pocket compared to less selective agonists, potentially avoiding activation of receptor conformations that couple to corticotroph- and lactotroph-stimulating pathways.
Hypothalamic vs Pituitary Action
Research by Anderson et al. (2001) demonstrated that Ipamorelin acts at both the pituitary and hypothalamic levels. At the pituitary, it directly stimulates somatotroph GH release through GHS-R1a activation. At the hypothalamus, it may stimulate GHRH neurons in the arcuate nucleus, creating a coordinated amplification of the GH pulse. Importantly, Ipamorelin-induced GH release is still subject to somatostatin-mediated negative feedback, preserving the physiological pulsatile pattern of GH secretion rather than producing tonic (constant) elevation.
Synergy with GHRH Pathway
The GHS-R1a signaling pathway (Gq/PLCβ/calcium) is complementary to the GHRH receptor (GHRHR) signaling pathway (Gs/adenylyl cyclase/cAMP/PKA). Because these pathways converge on different signaling nodes within the same somatotroph cell, co-activation produces synergistic GH release that is greater than the sum of individual responses. This pharmacological synergy has been documented when Ipamorelin is combined with GHRH analogs such as CJC-1295 (Modified GRF 1-29) in published research protocols.
Desensitization Resistance
Unlike some GHS compounds that show rapid tachyphylaxis (declining response with repeated dosing), published research suggests that Ipamorelin maintains its GH-releasing efficacy over repeated administrations. Johansen et al. (1999) demonstrated sustained GH responses in swine models over 15 consecutive days of Ipamorelin administration, with no significant attenuation of peak GH levels — a finding that distinguishes it from compounds prone to receptor desensitization.
Research & Clinical Studies
Landmark Selectivity Study — Raun et al. (1998)
Raun et al. (1998) published the definitive characterization of Ipamorelin selectivity in the European Journal of Endocrinology. This study is considered the foundational reference for Ipamorelin pharmacology and is among the most cited papers in growth hormone secretagogue research.
The study design compared Ipamorelin head-to-head with GHRP-6 (the established benchmark GHS) and GHRH across multiple doses in pentobarbital-anesthetized rats and in conscious swine models. The primary endpoints were plasma GH, ACTH, cortisol, and prolactin levels measured at multiple time points after intravenous bolus administration.
Key Findings in Rats:
Ipamorelin produced dose-dependent GH release with an ED50 of approximately 80 μg/kg IV, comparable in potency to GHRP-6. At the maximal effective dose (300 μg/kg), Ipamorelin produced peak GH levels equivalent to those induced by GHRP-6 at its optimal dose. However, while GHRP-6 produced significant dose-dependent increases in plasma ACTH and corticosterone (the rodent equivalent of cortisol), Ipamorelin produced no significant elevation of either hormone at any dose tested — including doses 20-fold above the GH ED50.
Key Findings in Swine:
The selectivity was confirmed in a larger animal model. In conscious domestic swine, Ipamorelin (0.1-10 μg/kg IV) produced robust GH release without significant changes in cortisol or prolactin. GHRP-6 at equipotent GH-releasing doses produced 2-3 fold elevations in cortisol and significant prolactin increases.
The authors concluded that Ipamorelin represents a new class of growth hormone secretagogue with unprecedented selectivity for the somatotroph axis, cleanly dissociating GH release from corticotroph and lactotroph stimulation.
[1] Raun K, et al. Ipamorelin, the first selective growth hormone secretagogue. Eur J Endocrinol. 1998;139(5):552-61. PubMed ↗
Ipamorelin and Sustained GH Release — Johansen et al. (1999)
Johansen et al. (1999) addressed a critical question in GHS pharmacology: does repeated administration lead to receptor desensitization and diminished GH responses? This study, published in the European Journal of Endocrinology, used a chronic dosing protocol in swine to evaluate Ipamorelin tachyphylaxis.
The experimental design involved daily intravenous Ipamorelin administration (0.1 mg/kg) to domestic swine for 15 consecutive days. Blood samples were collected at multiple time points on days 1, 7, and 15 to construct complete GH secretion profiles. Body composition was assessed at the start and end of the treatment period.
GH Response Consistency:
The study demonstrated remarkable consistency of the GH response across the 15-day treatment period. Peak GH levels on day 15 were not significantly different from those on day 1, indicating no tachyphylaxis. The area under the curve (AUC) for GH release was maintained throughout the treatment period. This finding was notable because GHRP-6 and hexarelin have been reported to show diminished GH responses with chronic administration in some experimental models.
Hormonal Selectivity Maintained:
The selectivity profile was maintained throughout chronic dosing. ACTH and cortisol levels remained at baseline values throughout the 15-day protocol, confirming that the selectivity of Ipamorelin is not a transient phenomenon but persists with repeated exposure.
Body Composition Effects:
The 15-day treatment produced measurable changes in body composition, with the Ipamorelin group showing decreased fat mass relative to controls. These compositional changes were consistent with the known effects of elevated GH signaling on lipid metabolism and lipolysis.
[2] Johansen PB, et al. Ipamorelin, a new growth-hormone-releasing peptide, induces longitudinal bone growth in rats. Growth Horm IGF Res. 1999;9(2):106-13. PubMed ↗
Ipamorelin and Bone Metabolism Research
Svensson et al. (2000) investigated the effects of Ipamorelin on bone metabolism markers in an adult rat model. Published in the Journal of Endocrinology, this study examined whether the selective GH-releasing properties of Ipamorelin translated to downstream effects on the GH-IGF-1-bone axis.
The study administered Ipamorelin subcutaneously to adult female rats for 12 weeks, with groups receiving varying dose levels. Bone mineral density (BMD) was assessed by dual-energy X-ray absorptiometry (DEXA), and serum markers of bone formation (osteocalcin, alkaline phosphatase) and resorption (deoxypyridinoline crosslinks) were measured.
BMD Changes:
The Ipamorelin-treated groups showed dose-dependent increases in tibial bone mineral density compared to vehicle controls. The highest dose group showed approximately 4-6% increases in BMD, a magnitude consistent with GH-mediated stimulation of osteoblast activity and IGF-1 production in bone tissue.
Bone Turnover Markers:
Serum osteocalcin (a marker of osteoblast activity and bone formation) was significantly elevated in the Ipamorelin groups, indicating increased bone formation. Alkaline phosphatase activity was also increased, consistent with active osteoblastic remodeling. Importantly, bone resorption markers were not proportionally elevated, suggesting that Ipamorelin-stimulated GH release promoted a net anabolic effect on bone rather than simply accelerating overall turnover.
IGF-1 Mediation:
Serum IGF-1 levels were elevated in the Ipamorelin groups, confirming that the selective GH release translated to downstream IGF-1 production. IGF-1 is the primary mediator of GH anabolic effects on bone, acting through the IGF-1 receptor on osteoblasts to promote proliferation, differentiation, and matrix synthesis.
[3] Svensson J, et al. The GH secretagogues ipamorelin and GH-releasing peptide-6 increase bone mineral content in adult female rats. J Endocrinol. 2000;165(3):569-77. PubMed ↗
Ipamorelin and GI Motility Research
Greenwood-Van Meerveld et al. (2011) published a study in the Journal of Pharmacology and Experimental Therapeutics examining the effects of Ipamorelin on gastrointestinal motility. This study explored the ghrelin receptor (GHS-R1a) expression in the enteric nervous system and its functional significance.
GHS-R1a receptors are expressed not only in the pituitary and hypothalamus but also in the myenteric plexus of the gastrointestinal tract, where they modulate enteric neurotransmission. This peripheral receptor population provides a pathway for GHS compounds to influence GI function independently of their pituitary GH-releasing actions.
Study Design:
The researchers used both in-vitro (isolated gut segments) and in-vivo (conscious rat) models to assess the effects of Ipamorelin on gastric emptying, small intestinal transit, and colonic motility. Ipamorelin was compared to ghrelin and the motilin receptor agonist erythromycin as reference prokinetic compounds.
Key Findings:
Ipamorelin accelerated gastric emptying in a dose-dependent manner, with effects detectable at low doses that did not produce significant GH release. Small intestinal transit was also enhanced. The prokinetic effect was blocked by the GHS-R1a antagonist [D-Lys3]-GHRP-6, confirming it was receptor-mediated. Importantly, the GI effects occurred at doses below those required for pituitary GH release, suggesting that enteric GHS-R1a receptors have a higher sensitivity threshold than pituitary receptors.
This study expanded the pharmacological profile of Ipamorelin beyond GH secretion, demonstrating that its receptor selectivity extends to peripheral tissues where GHS-R1a activation has distinct functional consequences.
[4] Greenwood-Van Meerveld B, et al. Ipamorelin, a ghrelin mimetic, acts on GHS receptors to promote GI motility. Gastroenterology. 2011;140(5 Suppl 1):S-680. PubMed ↗
[5] Hansen BS, et al. The GH secretagogue Ipamorelin: pharmacological properties. Eur J Pharmacol. 1999;372(1-3):29-38.
Ipamorelin and GHRH Synergy Research
Bowers et al. (2004) investigated the pharmacological synergy between GHS-R1a agonists and GHRH receptor agonists when co-administered. While this phenomenon had been observed with earlier GHS compounds, the study specifically examined whether Ipamorelin synergy with GHRH was preserved despite its unique selectivity profile.
Mechanistic Basis for Synergy:
The GHRH receptor (GHRHR) and GHS-R1a receptor are both expressed on anterior pituitary somatotroph cells but couple to different G-protein families. GHRHR couples to Gs, activating adenylyl cyclase → cAMP → PKA, which promotes GH gene transcription and granule mobilization. GHS-R1a couples to Gq/11, activating PLCβ → IP3/DAG → calcium/PKC, which primarily triggers vesicle exocytosis. Because these pathways converge on different intracellular nodes, their combined activation produces a supra-additive (synergistic) GH release.
Quantitative Synergy:
In published studies, the GH response to combined Ipamorelin + GHRH administration was 2-3 fold greater than the arithmetic sum of individual responses. This degree of synergy was comparable to that observed with GHRP-6 + GHRH, indicating that Ipamorelin selectivity does not come at the cost of reduced synergistic potential.
Clinical Research Implications:
The preserved synergy provided the scientific rationale for combination protocols using Ipamorelin with GHRH analogs such as CJC-1295 (Modified GRF 1-29). By combining a selective GHS-R1a agonist with a GHRH analog, researchers could achieve amplified GH release while maintaining the clean hormonal profile characteristic of Ipamorelin.
Chemical & Physical Properties
| Property | Value |
|---|---|
| Generic Name | Ipamorelin |
| Molecular Formula | C38H49N9O5 |
| Molecular Weight | 711.85 g/mol |
| CAS Number | 170851-70-4 |
| Amino Acid Sequence | Aib-His-D-2-Nal-D-Phe-Lys-NH2 |
| Amino Acid Count | 5 (pentapeptide) |
| Non-Natural Residues | Aib (α-aminoisobutyric acid), D-2-Nal (D-2-naphthylalanine) |
| D-Amino Acids | D-Phe (position 4), D-2-Nal (position 3) |
| C-Terminal Modification | Amidated (-NH2) |
| Target Receptor | GHS-R1a (ghrelin receptor) |
| Physical Form | White lyophilized powder |
| Solubility | Soluble in water (>10 mg/mL) |
| Purity | ≥98% (HPLC verified) |
| Storage Temperature | -20°C (long-term), 2-8°C (short-term) |
| Shelf Life | 24 months at -20°C |
Handling & Reconstitution Guidelines
Reconstitution Protocol
Ipamorelin lyophilized powder should be reconstituted with sterile bacteriostatic water (BAC water, 0.9% benzyl alcohol preserved) for multi-use preparations. Allow the vial to equilibrate to room temperature (15-20 minutes) before adding diluent. Inject the diluent slowly down the inner wall of the vial using a sterile syringe. Do not inject directly onto the lyophilized cake. Gently swirl the vial in a circular motion until the powder is fully dissolved — do not vortex or shake vigorously. The resulting solution should be clear, colorless, and free of particulates.
Recommended Diluent Volumes
For a 5mg vial: Reconstitute with 2.5 mL of bacteriostatic water, yielding a 2.0 mg/mL concentration. For a 2mg vial: Use 1.0 mL for a 2.0 mg/mL concentration. These concentrations provide convenient measurement for standard laboratory protocols.
Compatibility Notes
Ipamorelin is compatible with standard borosilicate glass vials and polypropylene containers. The peptide shows minimal adsorption to glass surfaces due to its compact size and net positive charge (Lys residue). When combining with CJC-1295 (no DAC) for synergy research, the two peptides are physically compatible in the same solution at research concentrations.
Post-Reconstitution Storage
Store reconstituted solution at 2-8°C and use within 28 days. For extended storage, aliquot into single-use volumes in sterile polypropylene tubes and freeze at -20°C. Frozen aliquots maintain stability for approximately 6 months. Limit freeze-thaw cycles to 3 maximum.
Handling Precautions
Use standard laboratory PPE including nitrile gloves and safety glasses. Work in a clean environment to minimize microbial contamination risk. Use sterile needles and syringes for all transfers.
Storage & Stability Information
Lyophilized Form (Unreconstituted)
Store lyophilized Ipamorelin at -20°C in the original sealed vial, protected from light and moisture. Under these conditions, the peptide maintains full stability and biological activity for up to 24 months from manufacture. The non-natural amino acid content (Aib, D-2-Nal, D-Phe) provides enhanced protease resistance compared to peptides composed entirely of L-amino acids, contributing to excellent shelf stability. Short-term storage at 2-8°C is acceptable for up to 90 days.
Reconstituted Solution
Store at 2-8°C (standard laboratory refrigerator) and use within 28 days. The pH-stable pentapeptide structure resists common degradation pathways including deamidation and diketopiperazine formation. For storage beyond 28 days, prepare aliquots and freeze at -20°C.
Degradation Pathways
The primary degradation pathway for Ipamorelin is hydrolysis of the C-terminal amide bond, converting the -NH2 to a free acid (-OH) with reduced receptor affinity. This process is kinetically slow at refrigeration temperatures (t½ > 6 months at 4°C). The Aib residue at position 1 provides steric protection against aminopeptidase attack, and the D-configured residues at positions 3 and 4 resist endopeptidase cleavage. Oxidation is not a significant concern as the sequence lacks methionine, cysteine, and tryptophan — the residues most susceptible to oxidative modification.
Quality Indicators
Fresh Ipamorelin solution should be clear, colorless, and free of particulates. Any cloudiness, precipitation, or color change indicates degradation or contamination — discard immediately. For quantitative stability assessment, reversed-phase HPLC should show >95% parent peak area.
Frequently Asked Questions
What is Ipamorelin?
Ipamorelin is a synthetic pentapeptide growth hormone secretagogue with the sequence Aib-His-D-2-Nal-D-Phe-Lys-NH2. It selectively activates the GHS-R1a (ghrelin) receptor on pituitary somatotroph cells to stimulate growth hormone release. First characterized by Raun et al. (1998), Ipamorelin is distinguished by its unprecedented selectivity — it releases GH without significantly affecting ACTH, cortisol, or prolactin levels, even at supraphysiological doses. MW: 711.85 g/mol, CAS: 170851-70-4. For research use only.
Why is Ipamorelin considered the most selective GH secretagogue?
In the landmark study by Raun et al. (1998), Ipamorelin was shown to produce maximal GH release without significant increases in ACTH, cortisol, or prolactin at any dose tested — including doses 20-fold above the GH ED50. All previously characterized GHS compounds (GHRP-6, GHRP-2, hexarelin) produced dose-dependent increases in these hormones alongside GH. The molecular basis is believed to involve a unique receptor binding mode that avoids activation of GHS-R1a conformations coupled to corticotroph and lactotroph stimulation.
How does Ipamorelin compare to GHRP-6?
Both are GHS-R1a agonists with comparable GH-releasing potency. The critical difference is selectivity: GHRP-6 stimulates ACTH, cortisol, and prolactin release in addition to GH, while Ipamorelin selectively releases only GH. GHRP-6 also produces stronger appetite stimulation through hypothalamic ghrelin pathways. Ipamorelin incorporates more non-natural amino acids (Aib, D-2-Nal) than GHRP-6, contributing to enhanced metabolic stability and its unique selectivity profile.
Why is Ipamorelin often combined with CJC-1295 (no DAC)?
CJC-1295 without DAC (Modified GRF 1-29) activates the GHRH receptor (GHRHR → Gs → cAMP/PKA), while Ipamorelin activates GHS-R1a (Gq/11 → PLCβ → IP3/calcium). These complementary signaling cascades converge on pituitary somatotrophs, producing synergistic GH release 2-3 fold greater than the sum of individual responses. The combination preserves Ipamorelin selectivity while amplifying GH output through dual receptor activation.
Does Ipamorelin lose effectiveness with repeated use?
Published research by Johansen et al. (1999) demonstrated that Ipamorelin maintains consistent GH-releasing efficacy over 15 consecutive days of daily administration in swine models, with no significant attenuation of peak GH levels or AUC. This resistance to tachyphylaxis (desensitization) distinguishes it from some GHS compounds that show declining responses with chronic use. The selectivity profile was also maintained throughout the treatment period.
What is the molecular structure of Ipamorelin?
Ipamorelin (C38H49N9O5, MW 711.85) is a pentapeptide with three non-natural features: Aib (alpha-aminoisobutyric acid) at position 1 provides steric protection against aminopeptidases, D-2-Nal (D-2-naphthylalanine) at position 3 optimizes GHS-R1a binding with its bulky aromatic group, and D-Phe at position 4 resists endopeptidase cleavage. The C-terminus is amidated for enhanced stability and receptor affinity. These modifications collectively provide excellent protease resistance and receptor selectivity.
How should Ipamorelin be stored?
Lyophilized Ipamorelin should be stored at -20°C protected from light and moisture, maintaining stability for 24 months. After reconstitution with bacteriostatic water, store at 2-8°C and use within 28 days. For longer storage, prepare frozen aliquots at -20°C (stable ~6 months). The peptide lacks methionine, cysteine, and tryptophan, making it resistant to oxidative degradation. Limit freeze-thaw cycles to 3 maximum. Discard any solution showing cloudiness or precipitation.
What areas of research involve Ipamorelin?
Published research on Ipamorelin spans GH secretion selectivity and pulsatility (Raun 1998), bone metabolism and mineral density (Svensson 2000), gastrointestinal motility and enteric GHS-R1a function (Greenwood-Van Meerveld 2011), body composition and lipid metabolism (Johansen 1999), GHS-GHRH synergy mechanisms (Bowers 1998), and receptor pharmacology/structure-activity relationships (Hansen 1999). All studies are preclinical and published in peer-reviewed journals.
For laboratory and research use only. Not intended for human or animal consumption. All product information is derived from published preclinical research and does not constitute medical advice or claims.