O Nascimento de uma Revolução: Como Peptídeos Vulneráveis Ganharam Força
A história dos peptídeos sintéticos começou com uma descoberta humilhante: cadeias lineares de aminoácidos L-naturais, mesmo sintetizadas com perfeição química, eram destruídas em minutos quando expostas a ambientes biológicos. Pesquisadores demonstraram que essas moléculas enfrentavam um trio implacável de obstáculos: proteases ubíquas que clivavam suas ligações (exopeptidases atacando os terminais, endopeptidases fragmentando ligações internas), clearance renal acelerado (peptídeos abaixo de aproximadamente 5 kDa atravessavam livremente a filtração glomerular), e conformações múltiplas que reduziam dramaticamente sua afinidade e seletividade de ligação. Com meia-vida circulatória medida em minutos, esses compostos pareciam destinados a permanecer apenas curiosidades laboratoriais.[1][2]
A transformação dessa realidade desalentadora em uma das classes farmacêuticas mais promissoras da atualidade representa uma das histórias de sucesso mais notáveis da química medicinal moderna. Modificações químicas estratégicas — desenvolvidas através de décadas de pesquisa sistemática — conseguiram engenheirar propriedades específicas nos peptídeos: resistência à proteólise, meia-vida circulatória estendida, biodisponibilidade aprimorada, restrição conformacional, detectabilidade e capacidade de conjugação com outras moléculas. O resultado dessa evolução pode ser observado em medicamentos como agonista de GLP-1 e agonista dual de GLP, onde modificações precisas transformaram moléculas com minutos de atividade em terapêuticas com dosagem semanal. Para compreender o contexto de síntese, consulte nosso guia de síntese e manufatura de peptídeos e o artigo sobre SPPS.
Domínios Terapêuticos I: Metabolismo e Diabetes - A Revolução da Lipidação
A Estratégia Revolucionária de Ligação à Albumina
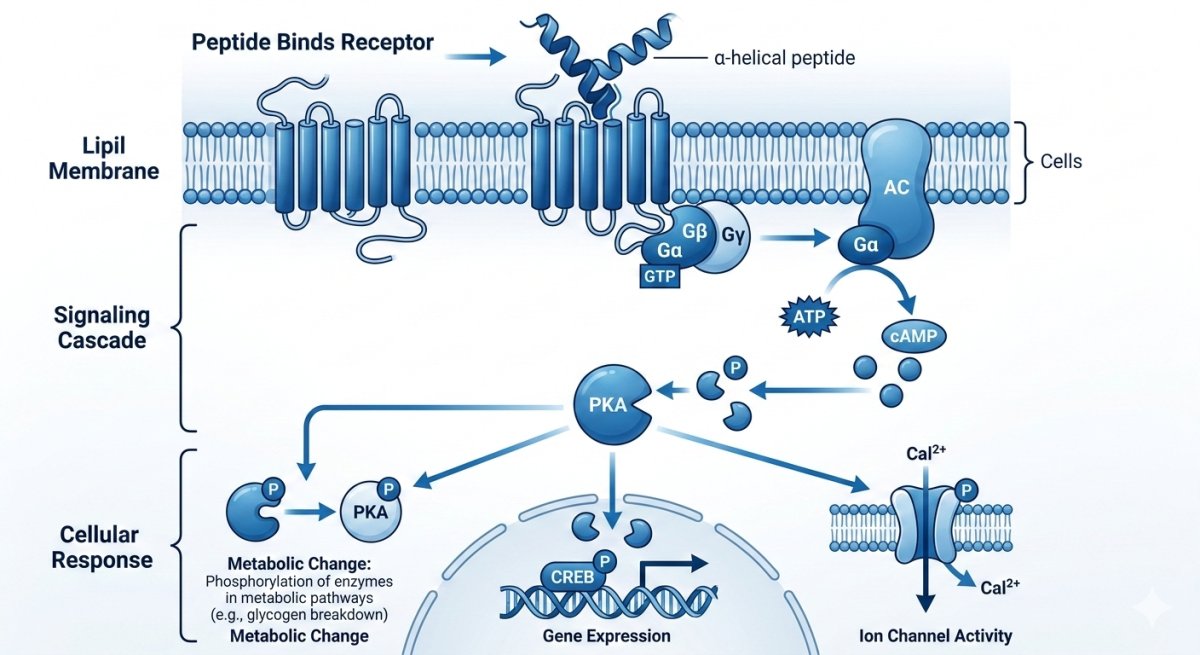
A lipidação — conjugação covalente de uma cadeia de ácido graxo ao peptídeo — emergiu como a modificação de maior impacto clínico na terapêutica peptídica contemporânea. O princípio subjacente demonstrou elegante simplicidade: uma cadeia de ácido graxo anexada ao peptídeo estabelece ligação não-covalente com albumina sérica (a proteína mais abundante no plasma sanguíneo, com meia-vida de aproximadamente 19 dias). Enquanto ligado à albumina, o peptídeo permanece protegido da filtração renal (o complexo albumina-peptídeo é demasiadamente grande para atravessar os poros glomerulares), blindado contra degradação proteásica (a superfície da albumina impede estericamente o acesso das proteases), e mantido em um reservatório circulante do qual o peptídeo se dissocia lentamente para exercer seus efeitos farmacológicos.[1][2]
agonista de GLP-1: O Paradigma do Diácido Graxo C-18
agonista de GLP-1 incorpora uma cadeia de diácido graxo C-18 (ácido octadecanodioico) conjugada através de um linker mini-PEG à lisina na posição 26 da sequência análoga ao GLP-1. Esta modificação lipídica específica foi engenheirada para alcançar ligação de alta afinidade à albumina mantendo concentração suficiente de peptídeo livre para ativação do receptor. O resultado é uma meia-vida circulatória de aproximadamente 165 horas (quase 7 dias), possibilitando injeção subcutânea semanal — comparado aos 1-2 minutos de meia-vida do GLP-1 nativo. A química do linker (um pequeno espaçador PEG entre a cadeia principal do peptídeo e o ácido graxo) proporciona flexibilidade conformacional que previne interferência da cadeia lipídica com a ligação ao receptor de GLP-1.[1][2]
agonista dual de GLP: Inovação com Diácido Graxo Insaturado C-20
agonista dual de GLP utiliza uma cadeia de diácido graxo insaturado C-20 (ácido eicosanodioico com uma única dupla ligação cis) anexada à lisina na posição 20, também através de um linker. A cadeia mais longa e a insaturação foram selecionadas para otimizar a afinidade de ligação à albumina e o perfil farmacocinético para a estrutura de agonista dual GIP/GLP-1. O posicionamento da conjugação lipídica em K20 (versus K26 na agonista de GLP-1) reflete a diferente cadeia principal peptídica — agonista dual de GLP é baseada na sequência GIP — e a necessidade de manter ligação tanto aos receptores GIP quanto GLP-1.[2]
Síntese de Peptídeos Lipidados
A lipidação pode ser realizada on-resin durante SPPS (através do acoplamento de um bloco de construção de ácido graxo pré-formado a uma cadeia lateral específica de lisina enquanto o peptídeo permanece ligado ao suporte sólido) ou pós-sinteticamente em solução (conjugando seletivamente o ácido graxo a um peptídeo purificado com lisina ortogonalmente desprotegida). A abordagem on-resin é mais eficiente para produção em escala de pesquisa, enquanto a conjugação em fase líquida pode ser preferida para escala de manufatura onde controle preciso do sítio de conjugação é crítico. Para qualquer abordagem, a lisina destinada à lipidação deve ser seletivamente desprotegida enquanto outras lisinas permanecem protegidas — estratégia possibilitada pelo uso de grupos protetores ortogonais (por exemplo, Dde ou ivDde para o sítio de lipidação, Boc para outras lisinas).[2][3]
Domínios Terapêuticos II: Tecnologia de Extensão Molecular - PEGuilação
A PEGuilação — conjugação covalente de cadeias de polietilenoglicol (PEG) a um peptídeo — foi a primeira estratégia amplamente utilizada para extensão de meia-vida e permanece importante para aplicações específicas. Cadeias PEG aumentam o raio hidrodinâmico do peptídeo (tornando-o demasiadamente grande para filtração renal), criam um escudo hidrofílico que reduz o acesso de proteases e reconhecimento imune, e melhoram a solubilidade de peptídeos hidrofóbicos em formulações aquosas.[1][2]
Pesos moleculares de PEG tipicamente variam de 2 kDa (para aumento modesto de tamanho) a 40 kDa (para extensão dramática de meia-vida). O trade-off é que cadeias PEG maiores progressivamente reduzem a afinidade de ligação ao receptor através de impedimento estérico da interação peptídeo-receptor — o chamado "dilema do PEG". PEGuilação sítio-específica (anexando PEG em posição definida distante da superfície de ligação ao receptor) mitiga esta questão mas requer design cuidadoso e química de conjugação ortogonal.
Embora a lipidação tenha largamente substituído a PEGuilação para terapêuticas baseadas em incretinas (a estratégia de ligação à albumina proporciona farmacocinética mais previsível com menor impacto na afinidade do receptor), a PEGuilação permanece amplamente utilizada para outras terapêuticas peptídicas e proteicas e para aplicações de pesquisa onde a cadeia PEG serve como marcador de detecção ou âncora de superfície.
Domínios Terapêuticos III: Estrutura e Função - Formação de Pontes Dissulfeto e Ciclização
Pontes Dissulfeto Intramoleculares
Muitos peptídeos bioativos requerem uma ponte dissulfeto entre resíduos de cisteína para integridade estrutural e função biológica. AOD-9604 contém uma ponte dissulfeto Cys183-Cys189 que restringe o peptídeo na conformação em alça requerida para sua atividade lipolítica. Ocitocina, vasopressina, somatostatina, insulina (com suas pontes dissulfeto intercadeias), e muitos peptídeos antimicrobianos dependem de pontes dissulfeto para sua estrutura tridimensional.[2][3]
Durante SPPS, cadeias laterais de cisteína são protegidas (tipicamente com grupos tritil em química Fmoc) para prevenir oxidação prematura. Após clivagem e desproteção global, os grupos tiol livres devem ser oxidados sob condições controladas para formar a ponte dissulfeto desejada. Para peptídeos com uma única ponte dissulfeto, oxidação ao ar em concentração diluída de peptídeo (0,1-0,5 mg/mL em tampão bicarbonato de amônio, pH 7,5-8,5) é frequentemente suficiente. As condições diluídas favorecem formação de dissulfeto intramolecular sobre intermolecular. DMSO (5-20% em tampão aquoso) acelera a oxidação mantendo seletividade para o produto intramolecular. Para orientação detalhada sobre estabilidade de pontes dissulfeto, consulte nosso artigo sobre estabilidade e armazenamento do AOD-9604.[3]
Múltiplas Pontes Dissulfeto
Peptídeos com duas ou mais pontes dissulfeto apresentam desafio regioquímico: oxidação aleatória de quatro ou mais resíduos de cisteína pode produzir múltiplos isômeros com diferentes padrões de pareamento dissulfeto, apenas um dos quais possui o dobramento nativo correto. Formação regiosseletiva de pontes dissulfeto utiliza grupos protetores ortogonais de cisteína — proteção diferente em cada par de cisteína que pode ser removida independentemente e sequencialmente. Por exemplo, um par pode usar proteção Acm (acetamidometil) enquanto outro usa Trt; o par protegido com Trt é desprotegido e oxidado primeiro, então o par Acm é desprotegido e oxidado em etapa separada, assegurando o padrão de pareamento correto.[3]
Ciclização Não-Dissulfeto
Ciclização através de métodos diferentes de pontes dissulfeto — incluindo ciclização de ligação amida cabeça-cauda (terminal N para C), pontes lactama entre cadeias laterais de lisina e glutamato/aspartato, e pontes tioéter (lantionina) — proporciona restrição conformacional sem a labilidade oxidativa das pontes dissulfeto. Peptídeos cíclicos geralmente exibem resistência à protease melhorada (ciclização remove terminais livres que são alvos primários de exopeptidases), seletividade de receptor aprimorada (conformação restrita reduz ligação a receptores off-target), e permeabilidade de membrana melhorada em alguns casos.[1]
Domínios Terapêuticos IV: Estabilização Terminal e Resistência Enzimática
Acetilação N-Terminal
Acetilação do grupo amino N-terminal (substituindo a amina livre por uma acetamida, Ac-) neutraliza a carga positiva no N-terminal e bloqueia degradação mediada por aminopeptidase dessa extremidade. É uma das modificações peptídicas mais simples e comumente aplicadas, realizada on-resin através do tratamento do N-terminal desprotegido com anidrido acético. Acetilação N-terminal também mimetiza mais proximamente o peptídeo como existiria dentro de um contexto proteico maior.[2]
Amidação C-Terminal
Amidação do grupo carboxil C-terminal (substituindo -COOH por -CONH₂) neutraliza a carga negativa no C-terminal e bloqueia degradação por carboxipeptidase. Amidação C-terminal é alcançada usando resina Rink amida (que libera o peptídeo como amida C-terminal mediante clivagem com TFA) ao invés de resina Wang (que produz o ácido livre). Muitos peptídeos bioativos naturais são C-terminalmente amidados in vivo pela monooxigenase alfa-amidante de peptidilglicina (PAM), e a forma amida é frequentemente requerida para atividade biológica completa.[2]
Domínios Terapêuticos V: Aminoácidos Não-Naturais - Engenharia de Resistência
Ácido Alfa-Aminoisobutírico (Aib)
Aib é um dos aminoácidos não-naturais mais amplamente utilizados no design de fármacos peptídicos. Seu carbono alfa possui dois grupos metil (ao invés de um metil e um hidrogênio como na alanina), criando restrição estérica que estabiliza conformação alfa-helicoidal e — criticamente — bloqueia acesso de protease a ligações peptídicas adjacentes. Tanto agonista de GLP-1 (Aib na posição 8) quanto agonista dual de GLP (Aib nas posições 2 e 13) incorporam Aib especificamente para conferir resistência à clivagem enzimática por DPP-4, que é a via de degradação primária para GLP-1 e GIP nativos. A substituição de tirosina no AOD-9604 segue o mesmo princípio de usar resíduo modificado para melhorar resistência à protease, embora através de mecanismo diferente.[1][2]
D-Aminoácidos
Incorporar D-aminoácidos (isômeros estereoquímicos espelho-imagem dos L-aminoácidos naturais) em posições específicas torna essas ligações peptídicas resistentes à clivagem por protease, porque a maioria das proteases são estereoespecíficas para substratos de L-aminoácidos. Substituição por D-aminoácidos pode estender dramaticamente a meia-vida peptídica em ambientes biológicos mantendo potencialmente ligação ao receptor se o sítio de substituição for tolerante à mudança estereoquímica. Peptídeos completamente com D-aminoácidos (análogos retro-inverso) são essencialmente invisíveis às proteases mas podem ter propriedades de ligação ao receptor alteradas.[1]
Aplicações de Pesquisa: Modificações Especializadas para Investigação Científica
Marcação Fluorescente
Conjugação de corantes fluorescentes (FITC, rodamina, Cy3, Cy5, série Alexa Fluor) a peptídeos possibilita visualização de localização peptídica, ligação ao receptor, internalização e tráfego em estudos celulares e teciduais. Fluoróforos são tipicamente conjugados ao N-terminal, uma cadeia lateral específica de lisina, ou um tiol de cisteína via química maleimida. A escolha do fluoróforo, sítio de conjugação e comprimento do linker podem todos afetar a atividade biológica do peptídeo — idealmente, o marcador deve ser posicionado em local que não participe da ligação ao receptor.[1]
Biotinilação
Conjugação com biotina possibilita detecção através da interação biotina-estreptavidina de afinidade extremamente alta (Kd aproximadamente 10⁻¹⁵ M). Peptídeos biotinilados são utilizados em ensaios de pull-down, imobilização de superfície (para estudos de ligação SPR ou BLI), detecção baseada em ELISA, e purificação por afinidade de parceiros de ligação. Biotina é tipicamente anexada ao N-terminal ou cadeia lateral de lisina através de espaçador PEG ou ácido aminohexanoico que posiciona a biotina distante da região ativa do peptídeo.
Marcação Isotópica
Peptídeos marcados com isótopos estáveis (incorporando aminoácidos marcados com ¹³C, ¹⁵N, ou deutério) servem como padrões internos em proteômica quantitativa baseada em espectrometria de massa. Esses peptídeos pesados possuem propriedades cromatográficas e de ionização idênticas às suas contrapartes naturais mas são distinguidos por mudança de massa definida, possibilitando quantificação absoluta de concentrações de peptídeos endógenos em amostras biológicas.
Tecnologia Avançada: Peptídeos Grampeados
Grampeamento peptídico — introdução de ponte de hidrocarboneto entre dois resíduos não-adjacentes usando química de metátese de olefina — restringe o peptídeo em conformação alfa-helicoidal. Peptídeos grampeados exibem resistência à protease dramaticamente melhorada (a cadeia principal restrita é substrato pobre para proteases), permeabilidade de membrana celular aprimorada (o grampo de hidrocarboneto aumenta hidrofobicidade geral), e afinidade ao alvo aumentada (a hélice pré-organizada paga penalidade entrópica menor mediante ligação). Esta tecnologia está sendo aplicada a peptídeos que visam interações proteína-proteína intracelulares — alvos historicamente considerados não-drogáveis.[1]
Implementação Estratégica: On-Resin versus Pós-Sintética
Modificações peptídicas se enquadram em duas categorias amplas de implementação. Modificações on-resin são realizadas durante SPPS enquanto o peptídeo permanece ligado ao suporte sólido — exemplos incluem acetilação N-terminal, incorporação de aminoácidos não-naturais (Aib, D-aminoácidos), lipidação on-resin, e marcação on-resin. A vantagem é que o peptídeo ligado à resina pode ser lavado para remover reagentes em excesso, e a modificação é incorporada ao fluxo de trabalho de síntese padrão. Modificações pós-sintéticas são realizadas no peptídeo clivado e purificado em solução — exemplos incluem formação de pontes dissulfeto, PEGuilação e lipidação em fase líquida, modificações enzimáticas, e algumas químicas de conjugação incompatíveis com condições on-resin. Modificações pós-sintéticas requerem etapas de purificação adicionais após conjugação para remover reagentes não reagidos e separar o produto modificado do material de partida não modificado.[2][3]
Perspectivas Futuras: A Nova Era dos Peptídeos Sintéticos
A evolução das modificações químicas transformou peptídeos de moléculas frágeis e de vida curta em ferramentas de pesquisa estáveis e terapêuticas de longa ação. Lipidação (possibilitando ligação à albumina para dosagem semanal de agonista de GLP-1 e agonista dual de GLP), PEGuilação (aumentando tamanho e reduzindo clearance), formação de pontes dissulfeto (restringindo conformações bioativas no AOD-9604 e muitos outros peptídeos), proteção terminal (bloqueando degradação por exopeptidase), incorporação de aminoácidos não-naturais (Aib para resistência a DPP-4), e marcadores específicos de pesquisa (fluoróforos, biotina, isótopos) cada um endereça limitações específicas do peptídeo não modificado.
Pesquisadores demonstraram que compreender o toolkit de modificações disponível — o que cada modificação realiza, como é sintetizada, e quais trade-offs introduz — possibilita aos cientistas projetar peptídeos otimizados para seus objetivos experimentais ou terapêuticos específicos. Esta revolução molecular continua evoluindo, com novas modificações sendo desenvolvidas para endereçar desafios emergentes na medicina de precisão e pesquisa biomédica avançada. O futuro dos peptídeos sintéticos promete ainda mais inovações, consolidando sua posição como uma das ferramentas mais versáteis e poderosas na arsenal terapêutico contemporâneo.